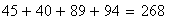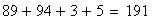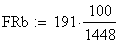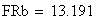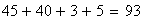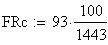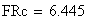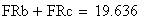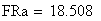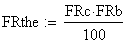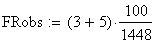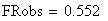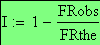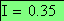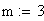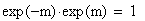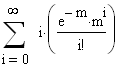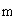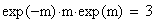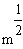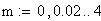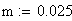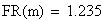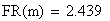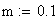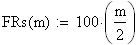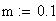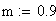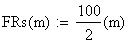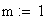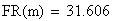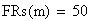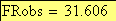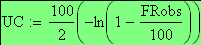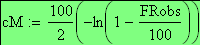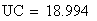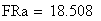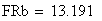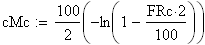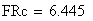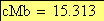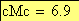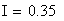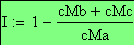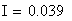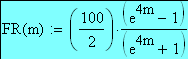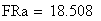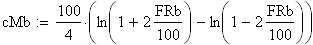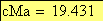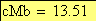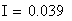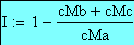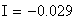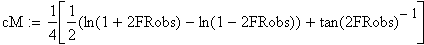Calculs des distances génétiques par analyse des recombinants
Crossing-over multiples - corrections des fréquences observées
Soit une croisement de deux parents P1 et P2 homozygotes pour trois charactčres et leurs allčles (dominant +) :
P1 +/+ y/y z/z donc phénotype : +yz
P2 x/x +/+ +/+ donc phénotype : x++
Les résultats de la F1 sont les suivants:
x + + 580
+ y z 592
-----------------
x y + 45
+ + z 40
-----------------
x y z 89
+ + + 94
-----------------
x + z 3
+ y z 5
-----------------
total 1448
a) examinons tout d'abord les combinaisons de x avec y nous avons:
b) examinons ensuite les combinaisons de x avec z nous avons:
c) examinons ensuite les combinaisons de y avec z nous avons:
On constate d'abord que les trois loci sont organisés selon un schéma:
x-----------------------------z---------------y
13.2% 6.4%
x----------------------------------------------y
18.5%
Nous constatons cependant que, alors que l'expression ci-dessous devrait donner 0, nous avons une erreur de 1.127
Les doubles recombinants sont au nombre de 8 soit une fréquence trop basse (0.552 au lieu de 0.85), en effet :
fréquence THEorique
fréquence OBServée
L'interférence I est définie comme suit:
I = 1 - OBS / THE
Si la fréquence des doubles recombinants est correcte alors I = 1-1 = 0, l'interférence est nulle
Si il n'y a pas de doubles recombinants alors I = 1- 0 = 1, l'interférence est maximale
-
Une conclusion vraisemblable est que la relation entre la fréquence et la distance n'est pas linéaire !!!!
-
Nous allons examiner une solution possible ŕ ce problčme, en faisant l'hypothčse suivante:
-
La distribution des événements de crossing-over suit, non pas une loi de probabilité égale, mais une loi de probabilité de type "distribution de Poisson".
-
Cette loi de distribution est applicables aux cas ou un événement trčs fréquent (un crossing-over) n'est échantillonné qu'avec de faibles fréquences (quelques dizaines de recombinants dans une expérience de génétique). Pour illustrer ceci de maničre plus concrčte: c'est la męme distribution que l'on obtient en pęchant des tétards avec une épuisette dans une mare oů ceux-ci sont abondants
Distibution de Poisson
i = nb de crossing-over
m = nombre moyen de crossing-over
m = le nb moyen d'événements (moyenne), c'est ŕ dire de crossing-over (CO) dans les classes d'entier : i 0,1,2,3,................................,Ą
Si dans une région du chromosome nous connaissons le nombre moyen de CO par meďose nous pouvons calculer la distribution (p) des meďoses ŕ (i) 0,1,2,3,........,Ą (i) CO.
p probabilité de meďose ŕ i CO en fonction de m; cette probabilité suit la loi de distribution de Poisson.
Nous pouvons utiliser les méthodes de calcul symbolique offertes par MathCad
se simplifie en:
et donne:
la moyenne d'un distribution de Poisson est :
se simplifie en:
et donne:
la déviation standard de la męme distribution est :
se simplifie en
et donne:
-
La seule classe qui nous intéresse est celle ou i = 0; en effet les meďoses au cours desquelles se produisent 1 ou plusieurs CO ( i > 0) se comportent de façon semblable, dans se sens que les fréquences de recombinaison parmi les gamčtes sont de 0.5 (50%).
-
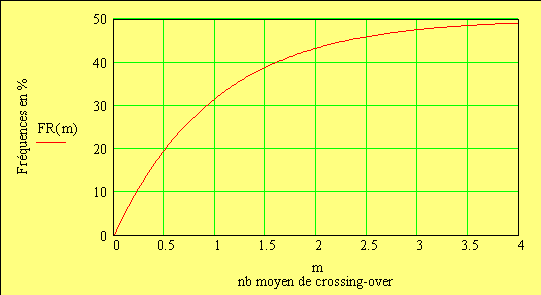
Le facteur déterminant les valeurs réelles de FR sera donc le rapport entre la FR de la classe 0 (i = 0) et celles des les autres classes. Les meďoses sans CO ont une FR de de 0 bien entendu. Examinons la situation pour m variant de 0 ŕ 4:
avec FR = fréquences de recombinaison
On remarquera que la courbe est presque linéaire pour des valeurs proches de 0
Nous pouvons dire que m est LA VRAIE mesure d'unité de cartographie génétique puisque m représente le nombre moyen REEL de crossing-over, alors que nous ne mesurons que la fréquence FR
La fonction peut se simplifier en :
La formule exacte reliant la fréquence ŕ m et i se simplifie lorque i = 0.
Nous avons maintenant une FONCTION CARTOGRAPHIQUE
et pour les petites valeurs de m (m < 0.1):
Un exemple de calcul est donné ci-dessous pour m = 0.5:
L'estimation la plus précise de la distance cartographique s'obtient en utilisant la fonction cartographique ou en cartographiant les gčnes par petits intervalles génétiques au sein desquels la relation entre FR et m est linéaire
La fonction inverse, dite de Haldane permet de calculer m ŕ partir de FRobs
Les unités cartographies UC ou cM sont égale ŕ m/2
en utilisant la FRobs mesurées dans l'expérience présenté plus haut nous avons:
Si nous calculons maintenant l'interférence I, ŕ partir des valeurs cartographiques en centi-Morgan, on constate qu'elle est négligeable
ŕ parti des fréquences brutes
ŕ partir des valeurs en cM
Voici maintenant l'allure, selon le modčle de Haldane, de la carte chromosomique pour les trois caractčres observés
x-----------------------------z---------------y
15.313 6.9
x----------------------------------------------y
23.115
Haldane, J. B. S. (1919). The mapping function. J. Genet. 8: 299-309.
Nous allons maintenant appliquer la fonction de Kosambi (1944). sans la démontrer; cette fonction est considérée comme plus précise que celle de Haldane
Un exemple de calcul est donné ci-dessous pour m = 0.5 (comme plus haut):
Kosambi, D. D. (1944). The estimation of map distances from recombination values. Ann. Eugenics 12: 172-175.
Voici maintenant l'allure, selon le modčle de Kosambi, de la carte chromosomique pour les trois caractčres observés
x-----------------------------z---------------y
13.51 6.481
x----------------------------------------------y
19.431
Si nous calculons maintenant l'interférence I, ŕ partir des valeurs cartographiques en centi-Morgan, on constate qu'elle est négligeable
ŕ parti des fréquences brutes
ŕ partir des valeurs en cM
Carter, T. C., and Falconer, D. S. (1951). Stocks for detecting linkage in the mouse and the theory of their design. J. Genet. 50: 307-323
http://web.rhul.ac.uk/Biological-Sciences/warren/index.html
http://www.ndsu.edu/ndsu/abergstr/webcourses/genetics/mf/mf09.htm
http://www.hgmp.mrc.ac.uk/Courses/Linkage/12.html
http://nitro.biosci.arizona.edu/courses/EEB320/EEB320.html